
Ever heard of the term bioinformatics? It’s an interdisciplinary field of science that combines different topics such as biology, computer science, mathematics and statistics. In a world where data is being generated at a faster rate than we can process, bioinformatics is used to analyze massive amounts of data and make sense of it all. We take a look at how bioinformatics is making a huge impact in drug discovery and design, by focusing on the field of molecular docking in virtual screening of compounds.
In Vivo, In Vitro, In Silico
You may have already heard of in vitro and in vivo; techniques that take place outside a living organism, and those performed within a living organism. A lesser-known third category of study, in silico, is the umbrella term for techniques that make use of computational power. The expression in silico was first used in 1989, meaning ‘performed on a computer or via computer simulation1.
When it comes to drug discovery, it is intuitive to see why in silico methods present good opportunities. The stages that follow the design of a new drug are both costly and time-consuming. The entire process of drug development can take from 12 to 15 years and cost billions of dollars, but in silico studies have been seen to both speed up the discovery rate and reduce (although not eliminate!) the need for expensive lab work.

In silico research is usually performed during the drug discovery stage, specifically lead identification in the medicinal chemistry phase. This step involves the use of many computational methods such as homology/comparative modeling, molecular docking, virtual high-throughput screening, quantitative structure-activity relationship methods (QSAR), conformational analysis and the list goes on.
In this article we will focus on molecular docking since the author has personally worked for some time with the technique!
What is Molecular Docking?
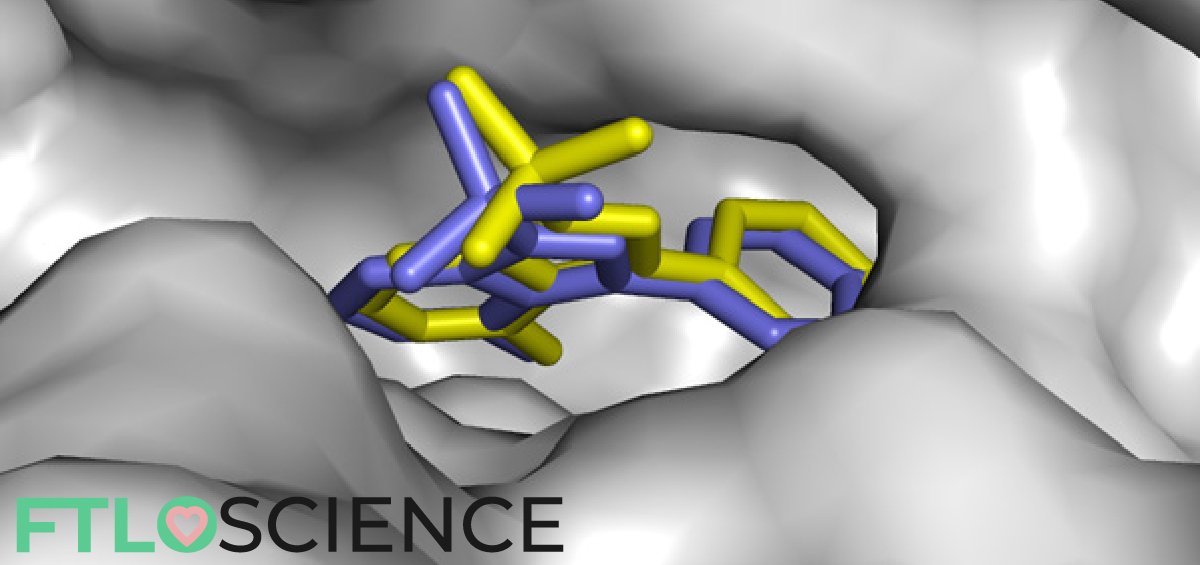
First off, let’s try to understand the concept of docking and its implications. Molecular docking is a type of bioinformatic modeling, an essential tool in structural molecular biology and in drug design. The purpose of using this technique is to predict the most likely ‘binding scenarios’ between a protein and a ligand, given their three-dimensional structures4,5.
Receptor-Ligand Binding
Receptors, such as G-protein-coupled receptors, are proteins found inside and on the surface of cells. They are responsible for virtually every single biochemical process inside our bodies. Collectively, molecules that bind to a receptor are called ligands. By binding, ligands can either activate receptors (agonists) or deactivate them (antagonists).
Being able to model the binding of receptors and ligands using molecular docking can be beneficial in the discovery of new drug targets or drug candidates.
If the 3D structure of the receptor is known, through X-ray crystallography, for example, it is possible to perform ‘docking’ simulations on it. Computational power can be used to predict–to a certain degree of accuracy–where and how well a given molecule can attach itself to the receptor.
Molecular Docking Using Computational Software
The diagram below shows a simplified depiction of how the docking procedure can influence and empower drug design. However, while this technique might seem to be able to reveal potential drugs rather easily, in silico methods and simulations are definitely not a substitute for good ol’ laboratory assays! The computational methods we have today are simply not advanced and robust enough to simulate the exact interactions between molecules, which grow exponentially with each atom involved.

The entire process is centered around using software to generate an enormous number of ligand-protein conformations, followed by calculations that predict which ones bind most strongly and are the most stable. The binding affinity of the ligand for the receptor is predicted by simulation software (there is a huge variety for both academic and commercial purposes), using mathematical equations known as scoring functions.
Scoring Functions
Scoring functions are used to predict the entropy or ‘binding free energy’ between the ligand and the target, a measure of their bond strength. With the aid of sampling algorithms and certain assumptions, they are a key part of molecular docking as we can attempt to reproduce the binding event as accurately as possible5,6.
The importance of the sampling algorithms used in docking procedures is evident when the sheer number of possible conformations between the ligand and protein is taken into consideration; factors that influence the binding energy include the huge number of translational, rotational and conformational degrees of freedom.
It turns out that this is actually way too much computation… even for a computer! In order to refine the search and save time and costs, these algorithms are put in place in order to remove improbable events.
Now that we’ve removed nonsense (or so we think) conformations that are unlikely to exist, we can sift through the manageable (but still enormous) pool of binding scenarios using the aforementioned scoring functions.
Rather than using precise calculations, these functions estimate the binding energies of many different conformations in a reasonable amount of time, albeit by sacrificing some degree of accuracy. The different scoring functions used are highlighted below:
Classical Force-Field functions
They assess the binding energy by calculating the sum of non-bonded interactions such as electrostatic interactions and Van der Waals forces. Some extensions of these calculations include other relevant aspects such as hydrogen bonds or entropy. It is important to take into account the solvent effects in these calculations, as it also plays a role in the free energy of the binding.
Empirical Functions
Empirical scoring functions use known binding affinities of protein-ligand complexes to perform multiple linear regression analyses. The values generated by this statistical model are then used as coefficients to adjust the equation in general. These ‘coefficients’ rely on the chosen dataset as well as the type of software used, making it common for differing results to be generated.
Knowledge-Based Functions
These functions use statistical analysis of ligand-protein crystal structures to obtain the measurement of the distance between them; it makes the assumption that an interaction that looks favorable will lead to activity. Its advantage lies in its computational simplicity, with a drawback being some interactions might not be represented in the available database of crystal structures. Using machine learning tools, the operator can also choose a protein set to ‘train’ the program with, producing different results.
This introduction provides a little bit of background behind the whole process of molecular docking; showing how software can calculate how well your drug can fit with the target protein.
Author’s Note:
Of course, there are other factors to take into consideration, such as how to treat individual receptors and ligands. Should they be considered rigid components that fit a certain position (an over-simplified version of the reality and because of that, not very popular nowadays) or as flexible structures?
I could keep going on and on with the whole process behind molecular docking, which is a big part of computing sciences, biology and even atomic theory, but I feel like I can stop here. Essentially, molecular docking is a highly versatile tool that provides a nice push to an overly-slow industry, making the development of new drugs a bit less tedious and definitely more interesting in terms of understanding how proteins bind!
Reference
- Mpkb.org. (2018). Differences between in vitro, in vivo, and in silico studies (MPKB). [online] Available at: https://mpkb.org/home/patients/assessing_literature/in_vitro_st/span>
- Yourgenome.org. (2018). How are drugs designed and developed?. [online] Available at: https://www.yourgenome.org/facts/how-are-drugs-designed-and-developed
- de Ruyck, J., Brysbaert, G., Blossey, R. and Lensink, M. (2016). Molecular docking as a popular tool in drug design, an in silico travel. Advances and Applications in Bioinformatics and Chemistry, Volume 9, pp.1-11.
- Gao, Q., Yang, L. and Zhu, Y. (2010). Pharmacophore Based Drug Design Approach as a Practical Process in Drug Discovery. Current Computer Aided-Drug Design, 6(1), pp.37-49.
- Hughes, J., Rees, S., Kalindjian, S. and Philpott, K. (2011). Principles of early drug discovery. British Journal of Pharmacology, 162(6), pp.1239-1249.
- Meng, X., Zhang, H., Mezei, M. and Cui, M. (2011). Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Current Computer Aided-Drug Design, 7(2), pp.146-157.
About the Author

Alejandra was a science writer at FTLOScience from October 2018 to April 2021.


