For a new drug entering the market in 2022, the costs behind its approval averages US$2 billion. In addition, the drug development process takes around 14 years of research and regulatory procedures before it is approved for sale. Despite this massive investment, pharmaceutical companies and research institutes continue developing new drugs to save and improve lives. We take a comprehensive look at the process and costs of drug development—how new drugs make their way from the lab bench to the pharmacy shelf.
Bringing a new drug to market in 2022 costs US$2 billion across 14 years of development. This process includes drug discovery, pre-clinical and clinical trials, regulatory filings and post-marketing surveillance.
(This article was first published in 2018. It has been updated in 2022 to account for new data, inflation and current FDA drug approvals.)
The Modern Drug Landscape
What’s often lost on people is how incredible it is that we find any human medicines at all. Every human being is 40 trillion cells, working together. I tell our people, you have to think every medicine we find is a miracle that fits in the palm of your hand. We’ve unlocked, in a sense, a billion years of evolution of the eukaryotic cell and human biology and somehow we found something able to move the needle in this incredibly complex system.
Vasant Narasimhan – CEO, Novartis
What are Drugs?
Drugs are humanity’s weapon against disease, providing patients with tangible improvements to their health and life expectancy. The U.S. Food and Drug Administration (FDA) recognizes any substance used in the diagnosis, cure, mitigation, treatment or prevention of disease as a drug.
This might sound simple, but developing a safe and effective medicine is complicated and expensive. Rigorous scientific research and science-based evidence are the foundations of drug development, separating modern medicine from ‘treatments’ such as aromatherapy and alkaline water.
To be effective, a drug must interact with a biological target such that it causes a beneficial change (such as binding to a receptor on a cell). Since our body is made up of tiny molecules, it was found that the proper dose of a specific molecule can exert these therapeutic effects.
Where Do Drugs Come From?
Such compounds can either arise from nature, through chemical synthesis, or any combination of the two. Contrary to popular belief, a compound made in nature is neither better nor safer than an identical one made in a lab. A molecule will behave like a molecule regardless of its origin.
Billions of years of evolution have enabled nature to produce compounds with highly complex structures, while lab-synthesized compounds are usually much simpler due to the limitations of the techniques available today.

For example, paclitaxel is an anticancer medication that comes from the bark of the Pacific yew tree. Aspirin, a commonly prescribed drug for pain and fever, can be synthesized in a laboratory.
Irrespective of their origin, these compounds have undoubtedly improved the standard of living in our species, protecting us from a variety of otherwise deadly diseases. Therapies like the antibiotic agent penicillin and vaccines like those against the smallpox virus are great examples of the enormous impacts of modern drug development on humanity.
Small Molecule vs. Biologic Drugs
Most medicines available today are ‘small molecule’ compounds, smaller chemicals that can be made from chemical reactions. There is another class of drugs called ‘biologic’ drugs that are made inside of living organisms, like bacteria.
Biologic drugs or ‘biologics’ include therapeutic proteins, monoclonal antibodies, immunotherapies, and even whole bacterial cells! Though there is no official chemical definition used to distinguish between small molecules and biologics, drugs that are produced using live cells instead of through synthetic chemical reactions are usually given the ‘biologic’ label.
These biologics present new and novel ways to tackle diseases. Complex treatments such as engineered immune cells can be infused into a patient, supplementing their immune system to fight diseases such as cancer.
Further improvements in biologic technologies are enabling the sector to target more diseases, creating biologic therapies that will likely eclipse the small molecule market in the future.
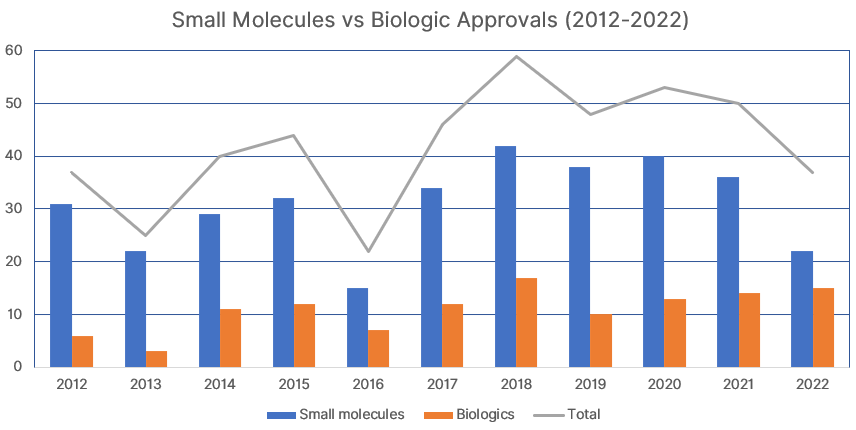
The graph above shows FDA drug approvals from 2012 to 2022, comparing between chemical entities (small molecules) and biologics approvals. In the last five years (2018-2022), 28% of drug approvals were for biologics, compared to 25% in the previous five years (2013-2017). 2022 was a record year for biologics, with a whopping 41% share of FDA new drug approvals that year.
There is no doubt that this increasing trend will continue in the coming years. Improvements to gene therapy and DNA engineering, along with Nobel prize-winning techniques like CRISPR are fueling the growth of biologic drugs.
How do Drugs Work?
In a drug, the compound that actually provides therapeutic benefit is known as the active ingredient, or active pharmaceutical ingredient (API). Most drugs contain the API and other compounds known as excipients. Excipients are inert components that help to support API release in the body, also making up the bulk of the drug by mass.
The API affects biological targets inside the body, resulting in changes to a particular disease or process. The actual chemical interaction is known as the mechanism of action, which can be complex. Even today, we do not have a complete understanding of many of the drugs on the market.
The effectiveness of a drug is measured by how well it exerts biological changes in the body; these signals are known as biomarkers. Choosing reliable biomarkers is important as they need to reflect the state of a disease in an observable manner.
In an anti-diabetic drug, for example, a good biomarker might be blood glucose levels in the patient. The drug is then only deemed to be effective if it causes blood glucose to drop by a certain amount, relative to placebo. However, many drugs work through a secondary action, such as controlling insulin levels in the body, which in turn affects blood glucose levels.
Due to the sheer number of similar processes in the body, drugs can also cause indirect and sometimes unrelated effects. A good clinical study will include the actual biological target as the primary biomarker, with indirect and adverse effects forming part of the observations.
Drug Discovery: Identifying a Drug Candidate
The drug industry harbors a dirty secret. New drugs are small organic molecules and small organic molecules are made by chemists.
Peter Goodfellow, Senior Vice President of Discovery Research at GlaxoSmithKline (Retired)
Drugs Begin as Molecules
For the purpose of this article, we will focus on small molecule drugs, as they still represent the majority of drugs on the market in 2022. These molecules are the starting point in drug discovery programs.
Pharmaceutical companies and research institutes maintain huge databases of molecules that have the potential to become drugs. They regularly screen these databases for ‘hits’, molecules that show activity toward a certain target.
This target is usually something that exists in our cells. G-protein coupled receptors, for example, are often physiological targets. But our body consists of over 40 trillion cells! It’s unfeasible to screen every compound for activity against all targets. How do we decide which molecules show promise?
Rational Drug Design
In the past, drug discovery was limited by the lack of structure elucidation techniques—many drugs were identified as the result of pure luck. Nowadays, visualization technologies such as X-ray crystallography, atomic force microscopy and mass spectrometry give us a better chance of rational drug design.
Rational drug design involves looking at structure-activity relationships. You see, interactions between molecules are determined only by their shapes (this is due to electronegativity and electron density).
This means that if we can establish a relationship between the structure of a molecule and its effects on a drug target, we can easily look for similar-shaped molecules! These structure-activity relationships help to identify compounds for screening.
Screening involves using high-throughput assays and computer programs to quickly determine if the chosen compounds have the potential to progress to the next stage of drug discovery. Generally, only the molecule’s activity against the target is measured at this stage.
Thousands of compounds can be screened at a time, producing many hits. Quantity is more important than quality at this point, since screening is a relatively inexpensive process. These hits are studied further in what is known as hit-to-lead optimization.
Hit-to-Lead Optimization
The hit compounds are then tested on more complex animal models or human tissue. This provides us with more data on each hit, such as its toxicity, pharmacokinetics and pharmacodynamics.
With this information, we can make modifications to the compounds to improve their safety and effectiveness. With every modification, the hits are tested and discarded if deemed too toxic or not effective enough, narrowing down the pool of hits to several lead compounds.
During hit-to-lead optimization, we can use existing structure-activity relationship data to help with making modifications to the hits.
For example, if there is a drug on the market for a specific disease, we can make small modifications to it to improve on its characteristics. Since the structure is similar, it is likely to have the same drug target. This is known the me-too approach in drug discovery.
Typically, several lead compounds are identified, but only one is chosen as the drug candidate. Scientists need to carefully choose the drug candidate from the pool of leads, because the next stage—development of the drug candidate—is an extremely time-consuming and costly process.
Intellectual Property (IP) Protection
There are two types of intellectual property (IP) protection that pharmaceutical companies receive: patent protection and market exclusivity. This acts as a form of insurance, ensuring that companies that invest in R&D get to profit from any potential successes.
Patent protection lasts for 20 years from the application date, and companies generally file for patents early on in the drug discovery process. This means that by the time the drug is approved, patents are usually close to expiry.
Another form of IP protection is market exclusivity, granted by the FDA after it approves the drug. During this exclusivity period, the pharmaceutical company that developed the drug has the sole rights for its manufacture and sale. The duration of market exclusivity is 5 years by default, but can vary between 0.5 years (generics) and 12.5 years (biologics).
Once both patent protection and market exclusivity expire, other manufacturers can produce ‘generic’ copies of the drug product from the now publicly available information. Generics are often much cheaper than their ‘brand name’ versions, but are essentially the same medicine.
Deciding on a Drug Candidate
Lead compounds usually have similar activity and safety profiles, which makes choosing a single drug candidate difficult. So how do pharmaceutical companies decide which compound is worthy of progressing to expensive pre-clinical and clinical trials?
An ideal drug candidate should also fulfill the following criteria:
- Have a well-defined pharmacophore: the active site of the molecule that interacts with the target. A pharmacophore is helpful for studying a drug’s mechanism of action; a clearly defined mechanism of action improves its chances of regulatory approval and patentability.
- Be easily modifiable. If a lead compound can be easily modified, its safety and efficacy can be further studied later. This essentially future-proofs the drug by providing an easy pathway for new drugs to be developed.
- Be safe and cost-effective during scale-up. The initial route of synthesis uses small-scale techniques in a laboratory. For a drug to be commercially viable, it needs to be suitable for large-scale manufacturing. Safety of the reactants is also a concern as hazardous chemicals can be expensive to store and protect against.
- Have simple reaction mechanisms. Chemists may be fond of fancy reagents and novel reactions, but from a manufacturing standpoint, the best chemistry is simple chemistry. Uncommon reagents are often costly. Well-established reactions with high yields that incorporate cheap catalysts under mild conditions are preferred.
- Adhere to green chemistry principles. Certain practices help improve the safety and reduce the environmental impact of chemical reactions. This applies to both small-scale lab experiments as well as large, industrial scales. These are explicitly stated in the 12 Principles of Green Chemistry.
- Be chemically stable during storage and transport. Ideally, drug products should be stable at room temperature and undergo limited degradation reactions (especially hydrolysis and oxidation).
Early drug discovery can be tricky, with twist and turns at every stage making the process heavily reliant on luck. The factors above are used to predict the chances of a drug candidate’s approval and feasibility to manufacture it at larger scales, but are by no means a guarantee of success.
Drug Development: Pre-Clinical and Clinical Trials
Ensuring a Safe and Effective Drug
Pharmaceutical companies and regulatory agencies must ensure that new therapies benefit the patient, preventing those that do not from entering the market.
For a drug candidate to become an approved drug, it needs only to fulfil two simple but strict requirements. It needs to be safe for patients, and it needs to be effective in treating its target disease.
Below is an overview of the drug development ‘funnel’, showing how potential new medicines are tested for safety and efficacy until only one FDA-approved drug remains.

The next stage of drug development—pre-clinical and clinical trials—is all about putting the drug candidate to the test, generating evidence to support its safety and efficacy.
Pre-Clinical Trials
Pre-clinical trials are the first step in the process, where qualified drug candidates are put to the test by studying their effects on animals. Lab rats are the model of choice; in addition to their ease of handling and anatomical similarity to humans, they are also relatively inexpensive.
Statistics show that our little rodent friends are test subjects in 95% of medical and pre-clinical studies1. They provide preliminary data on the safety and efficacy of the drug, as well as its mechanism of action and potential side effects.
However, precise data that is applicable to humans can only be obtained through the current gold standard of drug research: placebo-controlled, double-blind, human clinical studies.
In order to conduct such trials, companies must seek regulatory approval first, in the form of an IND application.
Formulation and Manufacture
Before submitting their drug for IND approval, pharmaceutical companies must ensure that the drug product is fully fleshed out. This means having an ingredient formulation that can successfully deliver the drug candidate. Apart from the active ingredient, most drugs also have non-active components known as excipients.
For example, in tablet manufacturing, excipients include fillers that form the bulk of the pill, binders that hold the ingredients together and coloring agents to help patients quickly identify the drug. Companies must also design the shape and size of their drug tablet as part of the formulation.
Companies must also have a manufacturing plan for their drug product. Apart from ensuring they have the capabilities to produce a stable supply of their drug, there must also adhere to strict manufacturing guidelines and regulations.
Different drug types require different pharmaceutical packaging; there are regulations on the suitability of certain materials. For example, some sterile injectable products must be packaged in type 1 glass, while oral liquid dosage forms can be stored in cheaper plastic containers.
The type of container used also affects the shelf life—the time it takes 10% of the active ingredient to degrade—of a drug, ideally between 1 to 5 years. For example, some plastics are permeable to water and air, which causes the degradation of drugs susceptible to hydrolysis and oxidation reactions. Glass containers can be colored using additives that protect its contents from photodegradation (light damage).
Drug products that are sold over-the-counter (OTC) in pharmacies are generally also required to have tamper-proof mechanisms in place.
IND Application
In the U.S., companies submit an Investigational New Drug (IND) application, presenting all the existing data on the drug to the Food and Drug Administration (FDA) The FDA will review this before granting approval, after which clinical trials can proceed.
The costs of drug development increases with each stage of the process. Hence, the pharmaceutical industry adopts a ‘fail fast, fail cheap’ approach to drug development of potential new medicines.
By the time of the IND application, companies can expect to have spent US$1 billion across 5 years of research. Although it sounds like a lot, the cost and duration goes up exponentially once the drug moves on to clinical trials, the next stage of development.
Hence, if the IND does not contain strong enough data, the company may decide to shelve the project and focus their efforts on developing a different drug candidate.
Phase One Clinical Trials
Phase 1 trials can begin once the FDA approves the IND. In these trials, a small group of healthy volunteers (usually 20 to 80) are invited to take very low doses of the drug to assess its metabolism pathways and overall safety. Close monitoring for safety data and undesirable side effects is a must; there have been cases of phase I trials causing unpredictable deaths.
As a rule of thumb, Phase I clinical trials of potentially life-saving drugs such as cancer therapies are almost always performed on actual patients rather than the usual healthy volunteers.
Phase Two Clinical Trials
Phase 2 trials continue with small-scale efficacy and dosing studies on a group of a hundred to a few hundred patients. The main goal of phase II trials is to determine the correct dosage that produces the desired therapeutic effect, with the dosing regimen finalized at this stage. Also collected is data concerning the safety and efficacy of the drug.
Phase Three Clinical Trials
Phase 3 trials involve a large number of patients (usually in the thousands). This allows for the efficacy of the drug to be fully established and compared with similar drugs already on the market.
To further ensure the drug’s viability, these studies usually encompass different populations and regions, different dosages, and even in combination with other drugs. Increasing the pool size enables the monitoring of side effects and adverse reactions under a range of different conditions.
The FDA usually requires at least 1 Phase 3 key trial to show favorable endpoints before the drug is approved.
Phase Four Post-Marketing Surveillance
But that’s not the end! Long-term studies (known as Phase 4 trials) continue even after the drug is approved. This post-marketing surveillance involves longer-term safety and tolerability studies of the drug, further adding to the costs of development.
The FDA regularly invites pharmaceutical companies to present regular safety updates from these trials to re-evaluate the status of the drug on the market.
In the past, phase IV trials have uncovered dangerous side effects in drugs, causing their withdrawal from the market.
On the other hand, these studies can also reveal new and often unexpected indications for the treatment of other diseases. This research strategy is known as drug repurposing.
The Entire Process: From Discovery to Market
Success Rates (Phase 1 to NDA Submission)
Following promising clinical data, the company then submits a New Drug Application (NDA). This dossier will include prior in vitro assays and in vivo animal studies, combined with human clinical trial data.
Also submitted is new knowledge from ongoing trials, such as information on drug metabolism and excretion.
Regulatory agencies like the FDA review all information—from manufacturing facility standards to the drug’s stability data to even checking for appropriate labeling information on the box—before choosing to approve it for sale on the market.
Average success rates of drug approvals by the FDA from 2009 to 20182:
- 13.8% of drug candidates that enter Phase 1 clinical trials are approved
- 35.1% of those that enter Phase 2 trials are approved
- 59.0% of those that enter Phase 3 trials are approved
- 83.2% of NDA submissions are approved
You can see that with each stage of the drug development process, the chances of success increases.
Success Rates (By Drug Type)
We know that just 13.8% of drugs that enter clinical trials are approved. The table below breaks this down further based on the therapeutic target of the drug candidates.
| Drug Candidate Type | Approval Rate |
|---|---|
| Oncology (antiancer drugs) | 3.4% |
| Endocrinology (hormones) | 19.6% |
| Cardiovascular (heart/circulatory system) | 25.5% |
| Central Nervous System (brain/spinal cord) | 15.0% |
| Autoimmune and Inflammation | 15.1% |
| Ophthalmology (eye disorders) | 32.6% |
| Infectious disease (including vaccines) | 25.2% |
| Other uncategorized | 20.9% |
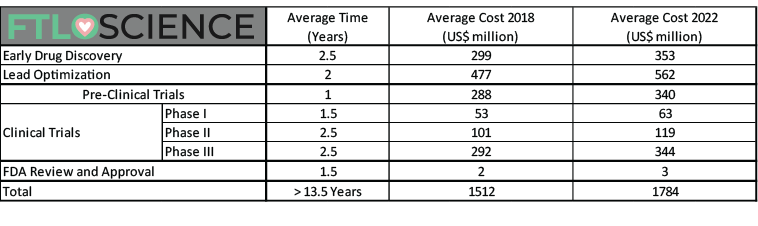
Total Costs of Drug Development (2022)
We can now calculate the staggering costs involved in the entire process of drug development, taking into account the cost of failed research based on a clinical trial success rate of 13.8%.
The table below shows the average time investment and costs of research and development for each approved drug. That is, how much a pharmaceutical company can expect to spend before they get a single new drug on the market.
The most updated figures are from 2018. An additional column has been added to adjust for inflation between 2018 and 2022. (Sources [1][2][3][4])
The figures above disregard Phase 4 post-marketing studies (US$204 million average).
In conclusion, a pharmaceutical company bringing a new drug to market in 2022 can expect to spend over US$2 billion across 14 years of development.
Is Pharmaceutical R&D Hindered by Costs?
Despite over $50 billion in yearly R&D spending by the larger pharmaceutical companies alone, the FDA only approves around 40 new chemical entities per year5.
Consequently, this severely impacts the industry and limits the potential for blue-sky discovery research. Uncertain returns on investment mean the pharmaceutical industry prefers to target already established markets.
However, the slow progress in discovering new and novel therapies is clearly not a problem of spending more. On average, the pharmaceutical industry invests more (% relative to sales) in research and development compared to any other manufacturing sector.
Compared to progress made in the semiconductor, electronics and even transportation industries, it is clear that pharmaceutical companies are somehow lagging behind despite the increasing costs of drug development.

The Future of Drug Development
Successes of Modern Drug Development
From the early days of penicillin to the complete eradication of the smallpox virus, we have proved time and time again that modern drug discovery and development are key to our progress as a civilization.
Once fatal, antiviral therapies have transformed HIV/AIDS into a now manageable (albeit chronic) disease. Thanks to new classes of drugs such as statins, mortality rates of cardiovascular diseases, coronary heart diseases and strokes have dramatically declined since 1950. And since peaking in the 90s, cancer-related deaths have fallen as well.

It is safe to say that drugs have played a big role in shaping society and humanity. But how long will this last?
A Worrying Trend
The current FDA approval rate for new drugs hovers at around 40 a year, and despite an increase in overall research expenditure, there doesn’t appear to be correlation between money spent and drugs being created.
Despite advances in our knowledge and cutting-edge technologies, it seems that we still do not have a clear picture of how to design and develop new and effective therapeutics!
Furthermore, pharmaceutical companies create a disproportionately high number of therapies for ‘Western’ diseases: coronary artery disease, stroke, lung cancers. There will always be a significant part of the population in need of a new cholesterol-lowering drug and more importantly, can afford to pay well for them.
Many ‘new’ therapies are simply improved versions of existing drugs (the ‘me-too’ approach) or the same drug used for a different purpose (drug repurposing). Can this be considered real innovation?
So why not follow in the footsteps of previous successes, such as Lipitor, a cholesterol-lowering therapy and the world’s all-time best-selling drug? The current model of drug development—considering the costs and drawbacks of true innovation—supports this approach. This is why there are always new drugs being developed to treat the same range of diseases.
Balancing Profit and Innovation
New treatments with novel mechanisms continue to have bigger impacts and bigger improvements in our lives, but it is a risky and expensive business for companies. Exciting fields like gene engineering have the potential to help us lead even healthier and longer lives. Pharmaceutical industry and regulatory agencies must work together to promote innovation in drug discovery and development, incentivizing and rewarding those who dare to venture.
The lack of breakthrough drugs is already a major cause for concern, especially in fields such as antibiotics and antifungals. Many pharmaceutical giants have also pulled the plug on their antibiotics R&D programs, leaving us vulnerable to future antibiotic-resistant bacterial strains.
Additionally, research into treatments for rare and tropical diseases is often neglected; this has to change. Rare diseases, as well as diseases prevalent in developing countries, affect millions but often do not receive the attention required. The FDA does have incentives in place to promote research into neglected diseases, such as the priority review voucher program. However, much more needs to be done to entice investment into drug development outside of ‘Western’ diseases.
In the meantime, pharmaceutical companies will continue to offset the huge costs of drug development by producing the same drugs for heart disease, strokes and diabetes, for which approval almost guarantees healthy returns.
Reference
- The Essential Need for Animals in Medical Research. (n.d.). Washington DC: Foundation For Biomedical Research.
- Wong, C. H., Siah, K. W., & Lo, A. W. (2018). Estimation of clinical trial success rates and related parameters. Biostatistics, 2018.
- Mestre-Ferrandiz, J., Sussex, J., & Towse, A. (2012). The R&D cost of a new medicine. Monographs.
- Paul, S. M., Mytelka, D. S., Dunwiddie, C. T., Persinger, C. C., Munos, B. H., Lindborg, S. R., & Schacht, A. L. (2010). How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nature reviews Drug discovery, 9(3), 203.
- Adams, C. P., & Brantner, V. V. (2006). Estimating the cost of new drug development: is it really $802 million?. Health Affairs, 25(2), 420-428.
- Kola, I., & Landis, J. (2004). Can the pharmaceutical industry reduce attrition rates?. Nature reviews Drug discovery, 3(8), 711.
- Dickson, M., & Gagnon, J. P. (2004). Key factors in the rising cost of new drug discovery and development. Nature reviews Drug discovery, 3(5), 417.
About the Author

Sean is a consultant for clients in the pharmaceutical industry and is an associate lecturer at La Trobe University, where unfortunate undergrads are subject to his ramblings on chemistry and pharmacology.