
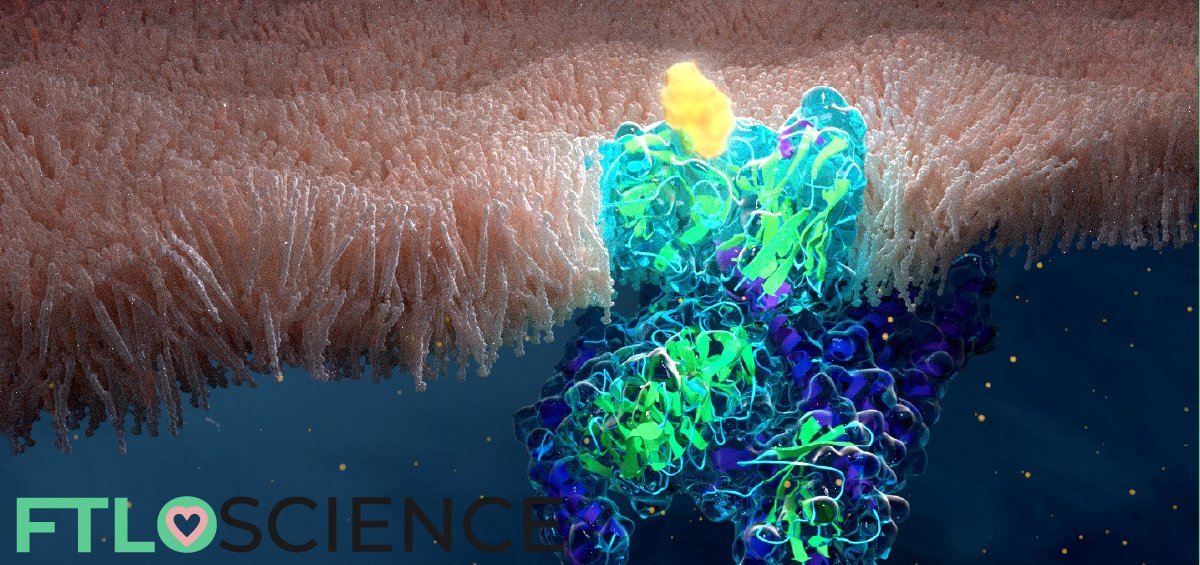
G-protein-coupled receptors (GPCRs) play a big role in the human body. As they maintain many of our key functions, much research is done to better understand the structure and function of GPCRs. In addition, GPCRs are potential starting points in many drug discovery programs. We look at how X-ray crystallography can provide a method to probe the structure of GPCRs, with a case study of the μ-opioid receptor.
Introduction to G-Protein-Coupled Receptors
G-protein-coupled receptors (GPCRs) control a variety of different processes in our body. They are found on the lining of cell membranes and help to transmit signals between the inside and outside of the cell. GPCRs respond to stimuli, ranging from chemical hormones, neurotransmitters, ions and even photons of light! This stimulus induces the receptor to perform some further biological functions.
As their name suggests, GPCRs contain G-proteins, also known as guanine-nucleotide-binding proteins. They sit inside on the part of the receptor inside the side, acting as molecular ‘switches’ and can turn other cellular components on or off. This, in turn, leads to a variety of responses. Characteristic of GPCRs are their 7 transmembrane (TM) loops, which span one side of the cell membrane to the other.
Collectively, molecules that bind to a receptor are called ligands. Ligands that activate receptors are known as agonists, while those that deactivate them are known as antagonists. GPCRs are involved in the control of almost every aspect of biological function, Understanding the structure-function relationships of GPCRs can, therefore, act as useful starting points in drug design and development.
X-Ray Crystallography
Protein X-ray crystallography and related techniques can produce high-resolution 3D structural images of large molecules such as receptors. A single pure crystal of pure protein scatters X-rays onto a detector, and rotation of the crystal provides a 3D scattering profile. Computational techniques then convert this scattering data into positions of the atoms, creating a 3D model of the protein.
Drug discovery and development programs rely heavily on X-ray crystallography to identify the structure of drug targets. The X-ray crystal structure of the human immunodeficiency virus (HIV) protease—an important enzyme in the virus’ lifecycle—was elucidated in the late 1980s. The 3D structure of the HIV protease provided structural information about its active sites, enabling us to predict and synthesize compounds that blocked them. This has led to the development of HIV protease inhibitors, potent drugs against HIV still in use today.

It might sound simple to ‘take a picture’ of a crystal, but the formation of a viable crystal of pure protein is another matter altogether. A sample of such purity can take months of optimization to obtain, following which crystallization must occur. Crystallization itself is largely trial and error because the conditions with favorable enthalpy are not immediately clear. Depending on the solvent system and the relative concentration of protein, it can take months or even years to form viable crystals.
Following this, data collection and processing is a tedious and time-consuming task, even with the help of supercomputers. Overall, the end-to-end process can take years, with high risks of failure throughout the entire project. Hence, the cost-to-benefit analysis of crystal structure elucidation is still a matter of much debate.
Case Study: The μ-Opioid Receptor
The μ-opioid receptor (μ pronounced ‘mew’!) can produce a range of effects in humans when activated by chemicals known as opioid alkaloids. Activation of the μ-opioid receptor (MOR) leads to an analgesic effect, making it an effective target for painkillers such as morphine. On the other hand, it also triggers mechanisms of tolerance and addiction.
Both natural and synthetic opioids exist. However, there is yet to be an agonist that activates the MOR in an optimal manner (i.e. one that provides good pain relief without the side effects of tolerance and addiction). The discovery of such a ligand will have potentially far-reaching healthcare, social and economic benefits1. Solving its structure might open up the possibility to create such an agonist. By conducting structure analysis, researchers can modify an existing compound such that it provides therapeutic benefit, while suppressing unwanted adverse effects.
Crystal Structure Elucidation
The mouse MOR was chosen as a starting point, sharing a 94% similarity with the human form. The poorly structured third intracellular loop on the μ-opioid receptor was identified as a hindrance to its crystallization due to its instability in solution. Hence, it was truncated and replaced with a more stable protein (T4 lysozyme) through GPCR engineering2.
Protein fusion techniques such as this are a common strategy in crystal engineering, serving to restrict the movement of transmembrane (TM) helices. Stabilizing these regions improves the chances of crystallization.
It was found that binding of the T4 lysozyme also improved polar (charged) interactions between the molecules, allowing for better crystallization. The protein crystals were produced within a lipid mesophase (a liquid-solid intermediate) solvent system, improving its solubility3.
Compared to traditional detergent solvents, a lipid mesophase also requires less solvent to be used to dissolve the protein, thereby reducing the time taken for a crystal to form.

The factors listed above provided an ideal environment for the crystallization of membrane proteins (including GPCRs) as they are generally unstable when removed from their native membrane environment. X-ray crystallography was then used to generate a scattering profile of pure mouse MOR protein fused with the T4 lysozyme, bound to an antagonist.
Elucidation of the first 3D crystal structure of MOR was reported by Kobilka’s group in 2012. Brian Kobilka won the Nobel Prize that same year for his work on G-protein-coupled receptors.
3D Structural Analysis
The μ-opioid receptor, like all GPCRs, is a membrane protein that consists of 7 linked alpha-helices with an extracellular N terminus and an intracellular C terminus. The topology of 7 linked alpha-helices is conserved across all GPCRs but may differ in the lengths of individual TM loops. The extracellular domains and ligand-binding regions are variable regions that differ between classes of GPCRs4.
The aforementioned alpha helices are situated within the lipid bilayer of the cell membrane, meaning they consist mainly of hydrophobic residues. They have to be aligned such that they all face outward while the remaining hydrophilic residues, if any, can form a polar core toward the center of the protein5. This polar region allows for charge interactions with incoming ligands, which is the case for MORs.
Studies on GPCRs show that when a ligand is bound, conformational changes to the transmembrane domains cause the entire three-dimensional structure of the receptor to change. This is what activates the G-protein signaling mechanism4.

The crystal structure of the MOR shows the binding interactions with a ligand, beta-funaltrexamine (β-FNA). It binds through at least 9 of its residues, with a total of 14 residues within 4 angstroms (Å) of the ligand. The nature of bonding consists of charge-charge interactions, hydrogen bonding, hydrophobic interactions, and a covalent bond also being formed between the K233 residue and a carbon atom on β-FNA. Also, 2 water molecules form a binding network with the H297 residue to interact with the phenol group on β-FNA1.
Future Drug Targets
The binding region of the μ-opioid receptor is situated close to the extracellular surface of the cell (as shown in Fig. 1a), forming a shallow groove. This suggests a loose binding interaction, with faster dissociation rates. Hence there is a higher possibility of the ligand being displaced by solvent molecules, deactivating the receptor.
In contrast, some receptor ligands are ‘buried’ deeper within the helical bundles and with their exit hindered by certain residues. Some muscarinic receptor ligands, for example, exhibit very slow dissociation kinetics1.
Due to the shallow binding region in MORs, ligands with few and/or weak interactions do not remain bound to the transmembrane helices for long. This also shortens the amount of time that the receptor can signal. Using structure-based design strategies to synthesize a ligand that binds more tightly, a more effective agonist can be produced to offset the ‘shallow’ binding region. This can be done, in theory, by increasing the strength or number of non-covalent interactions.
Other binding sites on MOR can also be exploited by interactions with ligands and possibly even biologic drugs. Adverse effects such as addiction is a common consequence of MOR activation, and there appear to be multiple sites on the receptor that can influence this6. Biologics may provide more specific targeting of certain sites on the receptor.
It is apparent that understanding the structure and function of the μ-opioid receptor (and other G-protein-coupled receptors) can be of great benefit. Furthermore, advances in recent fields such as bioinformatics mean we can take advantage of the vast amounts of data generated by such studies, converting it into information useful for drug discovery and development.
Cover graphic: artwork of a μ-opioid receptor on a cell membrane by Melanie (@nanoclustering)
Reference
- Manglik, A., Kruse, A. C., Kobilka, T. S., Thian, F. S., Mathiesen, J. M., Sunahara, R. K., … & Granier, S. (2012). Crystal structure of the ?-opioid receptor bound to a morphinan antagonist. Nature, 485(7398), 321.
- Rosenbaum, D. M., Cherezov, V., Hanson, M. A., Rasmussen, S. G., Thian, F. S., Kobilka, T. S., … & Kobilka, B. K. (2007). GPCR engineering yields high-resolution structural insights into beta-2-adrenergic receptor function. science, 318(5854), 1266-1273.
- Liu, W., & Cherezov, V. (2011). Crystallization of membrane proteins in lipidic mesophases. JoVE (Journal of Visualized Experiments), (49), e2501.
- Rosenbaum, D. M., Rasmussen, S. G., & Kobilka, B. K. (2009). The structure and function of G-protein-coupled receptors. Nature, 459(7245), 356.
- Crasto, C. J. (2010). Hydrophobicity profiles in G protein-coupled receptor transmembrane helical domains. Journal of Receptor, Ligand and Channel Research, 2010(3), 123.
- Contet, C., Kieffer, B. L., & Befort, K. (2004). Mu opioid receptor: a gateway to drug addiction. Current Opinion in Neurobiology, 14(3), 370-378.
About the Author

Sean is a consultant for clients in the pharmaceutical industry and is an associate lecturer at La Trobe University, where unfortunate undergrads are subject to his ramblings on chemistry and pharmacology.


