

At its core, the drug discovery process is remarkably straightforward: find a safe molecule with therapeutic benefits and turn it into medicine for patients. So why, then, do so many drug candidates fail? How can we improve early drug discovery, ensuring suitable drug candidates with better chances of successful clinical trial outcomes?
A molecule is ‘druggable’ if it has the potential to be the active ingredient in a drug product. The aim of drug discovery is to identify druggable molecules based on their activity, size, shape and functional groups present. However, conforming to these parameters does not guarantee a suitable drug candidate. Other early drug discovery strategies are required to improve the chances of a drug’s success.
An Overview of Early Drug Discovery
Screening: From Hit to Lead to Candidate
The first step in early drug discovery is identifying promising molecules, known as hits. Pharmaceutical companies and research institutions maintain vast libraries of molecules that they can quickly test for therapeutic efficacy using high-throughput screening assays. Molecules or proteins that show activity in these assays are known as ‘hits’.
Molecular hits are further narrowed down using secondary assays until a handful of ‘leads’ are left. These leads then move on to more thorough screens and animal models to test for their safety and efficacy. The most promising lead is labeled a ‘drug candidate’ and proceeds to clinical trials to test for safety and effectiveness in human patients.
Despite all these tests and screens, drug candidates have a poor success rate in clinical trials. Only 13.8% of drug candidates make it to regulatory approval, with certain drug classes like oncology only having an average success rate of 3.4%!
Challenges in Early Drug Discovery
Why is early drug discovery so inefficient in uncovering suitable drug candidates? Firstly, HTS techniques are designed for the rapid testing of many molecules. These screening assays filter the molecules to find hits to treat a particular disease, but they don’t test them against other possible physiological targets.
In the pharmaceutical industry, hits that are non-specific and show activity against other targets are generally poor drug candidates as they have higher chances of adverse side effects. However, these might only be apparent once the drug is tested in animal models or even human clinical trials.
Secondly, early drug discovery techniques cannot replicate the exact conditions within the human body (that consists of trillions of cells!) This leads to promising drug candidates failing to cross cell membranes or reach their intended targets, lowering their bioavailability in the patient to below therapeutic levels.
Finally, the financial side of drug development can present significant roadblocks. It takes, on average, US$2 billion over 14 years to create a drug from scratch, causing many medicinal chemistry programs to shut down at various stages due to a lack of funding.
Improving the Elusive Search for Suitable Drug Candidates
Looking Beyond Lipinski’s Rule of 5
To improve the chances of uncovering a molecule that might eventually become a blockbuster drug, pharmaceutical companies can employ several strategies, starting with a more robust investigation of what constitutes ‘drug-like’ properties.
In the past, it was thought that small molecules with good solubility and few binding sites were more likely to be ideal drug candidates, encapsulated in Lipinski’s Rule of 5:
- Molecular Weight < 500g per mole (small size)
- Log P value < 5 (water soluble)
- H-bond acceptors < 10
- H-bond donors < 5
The ‘Rule of 5’ is dead. Well, not really, but it has become increasingly clear that many drug candidates violate the traditional rules of early drug discovery. A study showed that more than 50% of drug molecules on the market actually fail to meet its criteria.
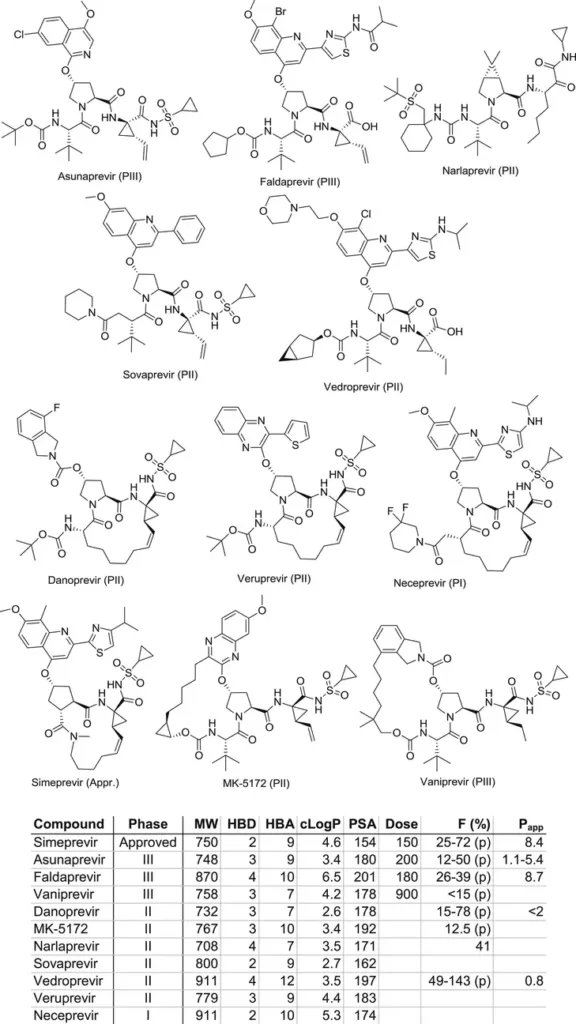
Small molecules have made way for larger, poorly absorbed biologic-derived molecules that virtually break every one of these rules. A record-high 41% of drugs approved by the FDA in 2022 were biologics, a trend that is on an upward trajectory.
A Robust Approach to High-Throughput Screening (HTS)
While we should not rule out hits that violate the Rule of 5, ensuring these molecules are safe and effective in patients is still the ultimate goal. That said, we can increase our chances of attaining this goal by employing HTS methods that look beyond potency toward a single therapeutic target. Data from multiple secondary targets can help paint a more physiologically relevant picture of the hit molecule.
We can further improve our understanding of the structure-activity relationship (SAR) between the molecular hit and the target with the help of visualization techniques such as X-ray crystallography and NMR. From a regulatory standpoint, robust SAR information increases the chances of a drug product being approved for the market.
Aside from increasing the variety of HTS techniques, the quality of screens must also be improved to represent biological conditions better. For example, the anti-fungal Posaconazole has a calculated log P value of 5.4, making it appear poorly water-soluble and un-druglike. However, experimental data shows it has a log P of 2.4, 1000 times more soluble than predicted!
An Early Focus on Formulation
Traditionally, pharmaceutical companies would identify a drug candidate as an active ingredient in a medication before developing a drug delivery formulation. The excipients (non-active ingredients) in the formulation help to release the active ingredient in the body but otherwise don’t influence the therapeutic activity.
However, many drugs fail in clinical trials due to the poor bioavailability of the active ingredient. This can be reduced by researching and modifying the drug formulation, especially for orally administered drugs. Changing the excipients and delivery method can often improve the bioavailability of a drug, leading to better outcomes in clinical trials and preventing delays.
An early focus on formulation is especially important today as we move away from small, soluble molecules. Active ingredients with poor solubilities often require specialized excipients to create oil-in-water dispersions or complexation with vehicles like cyclodextrins to become viable drug products. This might also require drastic changes in the formulation: tablet to soft gel capsules, oral to intravenous administration, etc.
It’s Cost-Effective to Fail (Early)
Ultimately, the drug development process presents a tradeoff, where early screening evaluates a large number of molecules with poor specificity, while the latter stages more thoroughly investigate a small number of compounds.
Of course, spending more time and money during the initial stages can increase the chances of developing a successful drug. However, failing early in the process is cheap, so quantity over quality is often the name of the game in early drug development. Pharmaceutical companies must be disciplined when planning their medicinal chemistry strategies, and know when to stick, twist, or pull out of projects altogether.
About the Author

Sean is a consultant for clients in the pharmaceutical industry and is an associate lecturer at La Trobe University, where unfortunate undergrads are subject to his ramblings on chemistry and pharmacology.


