

Stability testing of drug products is a regulatory requirement for pharmaceutical companies who want to market and sell their drugs. This article highlights the importance of stability testing and how stability programs are conducted. We will also look at several stability tests that are used to assess chemical degradation and determine the medicine’s overall shelf life.
The FDA requires stability testing data to be submitted for every drug product before approval and throughout its manufacture. Although specific test methods are not imposed, pharmaceutical companies must employ quantitative analysis (with upper and lower limits) to determine the drug’s chemical stability.
These quantitative methods include:
- Chromatography Assays
- Disintegration and Dissolution
- pH Analysis
- Moisture Analysis
- Microbial Content
Stability Studies in Drug Development
Why is Stability Important?
The stability of a medicine is important because it ensures the patient receives an effective amount of the drug molecule, along with its therapeutic effects. The stability of a drug product is affected by the following:
- Degradation of the ingredients
- Formulation stability
- Manufacturing process and environment
- Type of packaging containing it
- Conditions during storage and transport
In drug development, the stability of the drug product also determines its shelf life: the time it takes for 10% of the active ingredient to degrade. We can use stability study data to develop strategies for preventing degradation, thereby increasing a medicine’s shelf life.
It is also a regulatory requirement for pharmaceutical companies to submit stability testing data (including proposed shelf life) before they can market and sell their drug. Many regulatory agencies, including the U.S. FDA, also require post-authorization stability programs for quality control in manufacturing.
Long-Term Stability Programs
The long-term stability of a drug product is studied by storing it under typical conditions (different depending on the drug), listed below:
| Long-Term Studies | Temperature | Relative Humidity |
| General Storage | 25 °C | 60% |
| Refrigerator | 5 °C | Negligible |
| Freezer | -20 °C | Negligible |
Points to note:
- Pharmaceutical companies often use harsher conditions to cover long-term stability requirements in different parts of the world (up to 30 °C and 75% relative humidity).
- The humidity levels of refrigerator and freezer storage are negligible because there is little water vapor at those temperatures.
- If the storage material is made of plastic—a common material in pharmaceutical packaging—additional testing at lower relative humidity is required to study water loss from the drug product into the environment.
Chemical stability testing is performed every 3 months for the first year, every 6 months in the second year and then annually until the expiration date (usually less than 5 years).
The initial three batches (production cycles) of a drug product must undergo long-term stability programs to determine the shelf life of a drug. Subsequently, pharmaceutical companies must maintain a long-term stability program for at least one batch of their drug product every year.
Accelerated Testing
Drug products are often chemically stable over many years, which is excellent from a manufacturing point of view, but this also means that it can take an unfeasible amount of time before a drug experiences a significant degree of degradation.
With this in mind, the FDA allows for accelerated testing under harsher conditions to investigate the drug’s shelf life and degradation pathways. The results from these studies can be extrapolated using the Arrhenius equation to estimate the stability of the drug under normal storage conditions.
An example of accelerated testing conditions is shown in the table below. It is not recommended to use more extreme conditions as the calculated stability might not represent real-world data after correction.
| Accelerated Testing | Temperature | Relative Humidity |
| General Storage | Up to 40 °C | Up to 75% |
| Refrigerator | Up to 25 °C | Up to 60% |
| Freezer | Up to 25 °C | Up to 60% |
Stability Tests for Assessing Degradation
Chromatography Assays
Assays allow us to determine the relative amounts of specific chemicals in a mixture. In stability testing, the chemical of interest is usually the active ingredient. The gold standard for quantitative chemical assays is chromatography, in which chemicals in a drug product are separated and passed through a detector.
For polar (water-soluble) compounds, liquid chromatography is usually preferred. For non-polar (organic-soluble) compounds, we can use gas chromatography techniques.
A range of detectors can be used to perform quantitative assays, depending on the chemical of interest and the precision required. We then use the resulting chromatograms to calculate the relative concentrations of the different ingredients in a drug.
Many chromatography assays can also identify and quantify degradates of the active ingredient, which is helpful for identifying drug degradation pathways (oxidation, hydrolysis, photolysis, etc.). This information improves the chances of a drug candidate’s approval by regulatory agencies.
Some examples of detectors and their suitability for assaying different molecules are shown in the table below.
| Detector Type | Measured Parameter | Suitable Compounds |
|---|---|---|
| Mass Spectrometer | Ionized Atoms/Molecules | Universal |
| UV-Vis | UV-Visible Light Absorption | UV-Vis Active Molecules |
| Infrared | Infrared Light Absorption | IR-Active Molecules |
| Refractive Index | Light Refraction | Universal (Soluble) |
| Flame Ionization | Ion Flow | Hydrocarbons |
| Thermal Conductivity | Heat Conduction | Universal |
| Nitrogen-Phosphorous | Ion Flow | Nitrogen/Phosphorous-Rich |
For example, we can use a UV-Vis detector for proteins and large molecules as they tend to absorb light in the ultraviolet and visible spectrum. For small organic molecules, using a flame ionization detector might be more appropriate.
Disintegration and Dissolution Analysis
Solid oral dosage forms like tablets are designed to disintegrate and dissolve in our gastrointestinal tract. Most tablet formulations will undergo at least 70% dissolution (by mass) 45 minutes after administration.
However, degradation of the ingredients over time can alter a tablet’s dissolution rate, changing the speed at which the patient absorbs the drugs. Therefore, assigning acceptable limits for quality control is essential to confirm a drug product’s viability.
Stability testing for dissolution involves a disintegration tester that measures the time it takes for a tablet to disintegrate fully into particles (2000 μm/0.0787 inches or smaller).

Dissolution analysis is usually done by immersing a tablet into a physiological solution (0.1 M hydrochloric acid) at 37 °C with constant stirring to mimic the environment of the stomach and gastrointestinal tract.
Changes in pH of Liquid Drug Forms
Dissolved drug forms can alter their chemical structure over time, changing the pH of the overall liquid solution. Depending on the drug, the ingredients may be stable only within a specific pH range. pH changes also affect the solubility of dissolved solutes and can cause them to precipitate out of the solution.
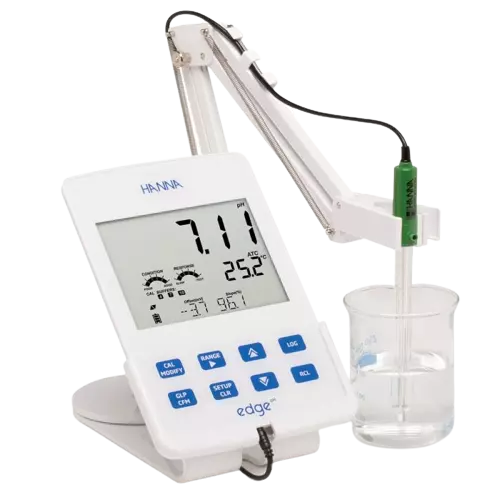
Stability testing for pH involves using a pH probe with several quality control measures in place:
- A calibration curve with at least 2 pH buffers that bracket the measured value
- Verification of the calibration with a solution of known pH
- Concurrent temperature readings at the point of pH measurement
Change in Water Content
Moisture is often a critical factor in degradation since many drug ingredients are susceptible to hydrolysis (chemical reaction with water). Drugs stored in moisture-permeable containers (especially some types of plastic) must also be monitored for water loss in dry environments, which can destabilize the drug product.
We can use moisture analysis to assess water content changes in solid and liquid drug forms. The industry standard test is Karl Fischer (KF) titration, where water in a sample is oxidized using sulfur dioxide and iodine into sulfuric acid and hydrogen iodide:
\text{2 H}_{2}\text{O} + \text{SO}_{2} + \text{I}_{2} → \text{H}_{2}\text{SO}_{4} +\text{ 2 HI}Once the endpoint is reached (iodine is in excess), the amount of iodine dispensed is used to calculate the amount of water in the sample. KF titration is often fully automated and can quickly calculate the moisture content for solid and liquid samples with high precision.

For quality control, the container needs to be suitably dry, as even small amounts of atmospheric moisture can give an inaccurate result. The apparatus must also be calibrated beforehand with a 1% water solution as a reference standard.
Microbial Content of Drug Samples
The last stability test required is to ensure the drug ingredients do not facilitate microbial growth over its shelf life. Since drug products are usually sealed after manufacture, there should be no significant microbial content from the beginning to the end of their shelf life.
To test for microbial content, the drug product can be incubated in an appropriate culture medium, and the number of colonies counted. Other tests for bacterial endotoxins can also test for the presence of pyrogens (toxic byproducts of specific bacterial strains).
Ensuring microbial quality is especially important for sterile medications since severe bacterial infections can result from administering contaminated drug products intravenously.
About the Author

Sean is a consultant for clients in the pharmaceutical industry and is an associate lecturer at La Trobe University, where unfortunate undergrads are subject to his ramblings on chemistry and pharmacology.


