How can we use artificial intelligence (AI) to improve drug discovery and development? In this installment of our deep dive, we walk you through how AI can accelerate primary and secondary screening during the drug discovery stage. We also introduce several startups using artificial intelligence and machine learning tools to create new medicines.
Drug Discovery: From Hit to Lead to Drug Candidate
Where Do Drug Molecules Come From?
Drug discovery is the process we use to discover new medications. Historically, drugs were mostly found purely by chance, such as by identifying the active ingredients in traditional medicines. Subsequently, classical pharmacology was used to investigate chemical libraries, including small molecules, natural products, or plant extracts and find those with therapeutic effects.
Seeking a new drug candidate in a chemical library often involves searching and wandering around in a vast chemical space. This ‘space’ comprises more than 1060 molecules, each waiting to be investigated by scientists (in case you need a comparison, there are ~1023 stars in our known Universe). Only a tiny fraction of these can realistically be studied, so how do we choose these select few?
To avoid having to sift through all 1060 compounds in the virtual chemical space, we can compromise by only searching for combinations of known therapeutically active ‘fragments’ or pieces in molecules (~1023 molecules). We can narrow this further by focusing only on the known chemical space. These are only chemicals that can be found on public databases and corporate collections—containing something like 108 molecules (still 100 million!)
Still, the known chemical space is far too ample for an exhaustive “exploration”. Scientists can only take partial, targeted peeks inside smaller virtual libraries and smaller chemical libraries (for example, PubChem, ChemSpider, ZINC, NCI, ChemDB, BindingDB, ChemBank, ChEMBL, CTD, HMDB, SMPDB, DrugBank). It is within these limited spaces that drug discovery begins.
Screening the Chemical Space for Hits
Numerous in silico methods are used to virtually screen compounds from various virtual chemical spaces and in vitro high-throughput screening experiments of chemical libraries (known chemical spaces) to identify novel drug molecules.
Once a suitable hit (a promising drug compound) is identified, a critical step in drug discovery is conducting preliminary drug metabolism and pharmacokinetics studies on the molecule, often referred to as ADMET (Absorption, Distribution, Metabolism, Elimination & Toxicity) studies.
Basic safety and efficacy data can be obtained from cell-based in vitro assays, animal studies, and recently: AI tools like the DeepTox algorithm, DeepNeuralNet-QSAR, Hit Dexter etc. (we will talk more about these later). It is important to note that rigorous testing is not done on hit compounds, as they are usually too many (quantity is the name of the game at this point).
At this point, researchers also evaluate hits based on their ease of synthesis. They explore different chemical pathways available to create the molecule, discarding the hits that require dangerous precursors or have poor atom economy (i.e. are not ‘Green‘).
Turning Hits into Leads, Leads into Drug Candidates
Following this, hits are discarded until several lead compounds remain. Lead compounds enter the drug design phase of the drug discovery process. This involves optimizing and modifying lead molecules to achieve a shape or charge that will better interact with a bio-molecular target in a (hopefully) therapeutic manner. It is important to note that the process between hit and lead is often fluid, with leads discarded and hits revived as more information is uncovered.
At the end of this process, researchers will evaluate all the data and bring forward the most promising lead compound to become a drug candidate. The drug candidate will progress into the drug development stage. This includes optimizing the manufacturing process for scaling up its synthesis and formulation and routes of administration studies.

To make a long story short, the drug discovery process involves:
- in silico studies that, in combination with cellular functional tests, are used to provide more information on the viability of compounds for their therapeutic activity
- screening hits (high throughput screening in vitro and secondary assays)
- medicinal chemistry (optimizing the molecule’s design and synthesis)
- optimization of hits to reduce potential drug side effects while increasing affinity and selectivity
What Role Does AI Play in Drug Discovery?
AI-based methods are increasingly being used throughout the drug discovery process, which can be divided into four major stages: target selection, target validation, compound screening and lead optimization.
For example, AI tools can be used in the following ways:
- predicting the 3D structure of a target protein
- designing new molecules
- quantum mechanics calculations of compound properties
- computer-aided organic synthesis
- real-time image-based cell sorting
- cell classification
- developing assays
In this article, we will focus on the different AI tools used in primary and secondary screening and introduce several startups that are paving the way for implementing AI in drug discovery.
AI in Primary Drug Screening
High-Throughput (Primary) vs. Secondary Screening
Discovering a new drug for a particular disease starts with high-throughput screening (HTS), which involves testing large libraries of chemicals on proteins, cells or animal embryos. HTS is done on microplates that can screen many compounds at once for their ability to modify a target.
During this process, we can quickly recognize active compounds that can modulate a particular bio-molecular pathway to achieve therapeutic benefit. For example, HTS robots can currently automate the testing of up to 100,000 compounds daily.
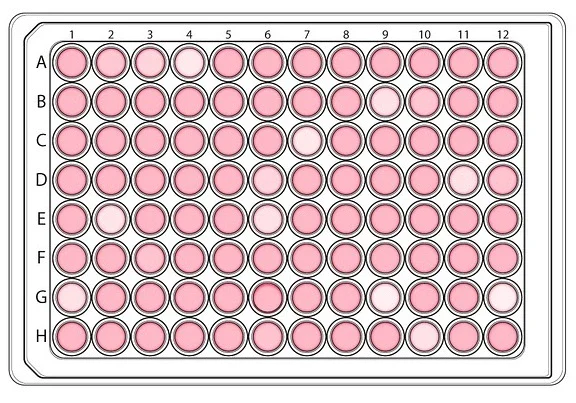
Drug screening is divided into primary screening, which allows high throughput measurements on cells (or proteins or embryos) of the compounds of a chemical library, and secondary screening (which we will discuss later in this article), which helps confirm the efficacy of primary screening results, by a series of functional cellular assays.
AI in Image-Based Profiling
During primary screening, specific AI tools are primarily used for sorting and classifying cells by image analysis (image-based profiling). In particular, for image-based phenotyping after treatment, we can use two approaches:
- The first approach involves screening applications (often also called high-content) focusing on a specific phenotype (a process in our cells, e.g., cell division). Phenotyping aims to identify drugs that can modulate function. For example, identifying a molecule that can inhibit tumor cell division/cell cycle.
- The second application of high-throughput imaging encompasses global profiling of all cellular changes after treatment and is complementary to techniques like transcriptomics, proteomics, metabolomics etc. To achieve this, the sub-cellular structures or specific proteins (relevant to a target process like cell division) are stained to ‘paint’ and visualize them. At the same time, automated image analysis is used to profile the changes to the cell during treatment.
In general, for this part of primary cell sorting and identification during screening, computer vision can extract multivariate feature vectors of cell morphology, such as cell size, shape, texture and staining intensity, without further human intervention, before and after treatment.
Specifically, all large-scale studies employ segmentation approaches — cell images are segmented from the background by varying the image contrast—to define cellular outlines before feature extraction accurately. Feature extraction is a method of capturing visual content of images for indexing using general features, such as extraction of color, texture and shape or domain-specific features.
AI Tools in Primary Screening
We must remember that AI tools are simply computer programs that follow a set of pre-programmed instructions. Therefore, it is vital to ensure the computer ‘sees’ what the human eye sees when we look at cells through a microscope.
To make this digital leap, we can obtain Tamura texture features from molecules during screening. The mathematical definitions of six original Tamura texture features are coarseness, contrast, directionality, line-likeness, regularity and roughness.
We can also extract wavelet-based texture features (texture analysis based on wavelet transformations) from each cell image. The principal component analysis (PCA) technique is used to reduce the dimensionality of the extracted features.
The AI tools are then trained to classify the different cell types. One of the most commonly used is the least-square support vector machine (LS-SVM) method—a set of related supervised learning methods that analyze data and recognize patterns—that shows the highest classification accuracy.
During screening, the AI-based image analysis decision-making must rapidly and accurately identify and classify different cell types during treatment. Accordingly, most modern image-activated cell sorting devices measure optical, electrical and mechanical cell properties quickly.
AI must match the speed of these instruments, allowing for flexible and scalable cell sorting automation, high-speed digital image processing and decision-making within a few tens of milliseconds.
AI in Secondary Drug Screening
Selecting High-Quality Hits Through Secondary Screening
Secondary screening aims to generate added information to transition from a relatively large number of possible hits generated during the primary screening to a few high-quality compounds that eventually will become lead candidates.
The process of secondary screening—also named focused or knowledge-based screening—involves selecting from the chemical library smaller subsets of molecules that are likely to have activity at the target protein, based on knowledge and literature. For example:
- biochemical — kinase/ATPase assays, protease assays, protein interaction assays
- cell-based — reporter assays, viability assays, GPCR and ion channel assays, qPCR.
Knowledge-based screening uses pharmacophores (a pharmacophore is a molecular arrangement of chemical features known to induce biological activity) and molecular modeling to understand the lead molecule better. In turn, this has given rise to virtual screenings of compound databases.
Before we see how AI and machine learning (ML) can help us during secondary screening, let’s first talk briefly about AI and ML.
What’s the difference between Artificial Intelligence (AI) vs. Machine Learning (ML)?
AI is the overarching technology that enables a machine to simulate human behavior and create intelligent systems following a set of rules. For example, AI can mimic human thinking and behavior by sorting objects based on their color.
ML is an application or a subset of AI in which the machine learns and alters the preset rules based on new input. This allows a computer to automatically learn from past data without being programmed explicitly. AI and ML are part of computer science and are often used in correlation.
Scientists can program a computer to think like us and perform tasks as a ‘normal’ human would—this is AI. They can then use ML algorithms (like reinforcement learning algorithms and deep learning neural networks) to develop and improve upon this’ intelligence’. The advantage of this is that the outcome may go beyond what a human can come up with!
For instance, a novel deep-learning algorithm (a type of ML that uses interconnected nodes or neurons in a layered structure resembling the human brain) called CeCILE (Cell Classification and In-vitro Lifecycle Evaluation) was developed in 2021 to study how cells react to radiation.
CeCILE can detect and analyze cells using videos from phase-contrast microscopy up to a sample size of 100 cells per image frame. Using an existing dataset as a ‘training’ model, CiCEILE provided more information about cell numbers, cell divisions and cell deaths over time than what was extracted by hand.
For those interested, the global cell-based assay and high-content screening market research for 2022-2023 are reviewed here.
AI Tools for Assessing Toxicity
An ideal absorption, distribution, metabolism, excretion and toxicity (ADMET) profiles are vital indicators of a drug’s success. The toxicology profile of a compound is arguably the most important of these parameters during the screening stage, as any toxic compounds can be quickly removed from the program.
An AI solution to tackle this exists in the DeepTox algorithm, which can first study the chemical structure of the compounds, process their potential toxicities and finally use this as the input for ML methods.
This input is known as a molecular descriptor and is the final result of a logical and mathematical procedure. Basically, the descriptor takes chemical information encoded within a molecular structure and transforms it into a helpful number based on a standardized experiment .
These descriptors are categorized into two groups:
- Static descriptors include atom counts, surface areas and the presence or absence of toxicophores in a compound. A toxicophore is a chemical structure or a portion of a structure that is related to a chemical’s toxic properties; there are around 2500 known toxicophores. Other chemical features extracted from standard molecular fingerprint descriptors are also calculated.
- Dynamic descriptors are calculated in a pre-specified manner according to the researcher’s input. Dynamic descriptor algorithms keep the dataset within manageable limits despite a potentially infinite number of different dynamic features.
So far, in typical test cases, the DeepTox algorithm has shown good accuracy in predicting the toxicology of compounds.
AI Tools for QSAR Models
Quantitative Structure-Activity Relationship (QSAR) studies attempt to build mathematical models relating a compound’s physical and chemical properties to its shape and structure. Such mathematical models can inform pharmacological studies by providing an in silico methodology to test or rank new compounds for desired properties without actual wet lab experiments.
These QSAR-based models have proven to be very valuable in predicting physicochemical properties, biological activity, toxicity, chemical reactivity and metabolism of chemical compounds during screening. They are now increasingly being accepted within the regulatory decision-making process as an alternative to animal tests for toxicity screening of chemicals.
An example of an AI tool for QSAR studies is the new MRA Toolbox v.1.0, a web-based toolbox for predicting the mixture toxicity of chemical substances in chemical products. This toolbox is a novel web-based platform including four additive toxicity modules: two conventional (Concentration Addition and Independent Action) and two advanced (Generalised Concentration Addition and QSAR–based Two-Stage Prediction) models.
Several Deep Neural Networks (DNNs)—complex computational models such as computer vision and natural language processing— generated similarly promising results for QSAR tasks (DeepNeuralNet-QSAR).
Previous work showed that DNNs could routinely make better predictions than traditional methods, such as random forests, on a diverse collection of QSAR data sets. It was also found that multitask DNN models—those trained on and predicting multiple QSAR properties simultaneously—outperform DNNs trained separately on the individual data sets in many, but not all, tasks.
Finally, QSOR (Quantitative Structure Odor Relationship) modeling is a subdomain of the QSAR, with more complex algorithms in deep learning and neural networks for predicting the odor of a compound based on molecular structure alone.
Machine Learning Tools for Secondary Screening
MMPs (matched molecular pairs) are closely related molecules that only differ by a single atom or functional group; hence, we predict them to have similar physicochemical characteristics. MMP analysis involves comparing these pairs to learn more about the drug molecule and the target site.
A typical QSAR study generates MMPs for de novo design tasks. De novo (“from the beginning”) drug design means taking a purely computational approach, where a molecule is virtually constructed, atom by atom, based on the active site of a target receptor protein.
An MMP analysis can then investigate how a single localized change to the molecule will impact its properties and bioactivity as a drug candidate.
There are currently three ML methods available for MMP analysis:
- the Random Forest (RF), a classification algorithm consisting of many decisions trees
- the Gradient Boosting Machines (GBMs), an ML technique for regression and classification problems
- the Deep Neural Networks (DNNs) that were previously applied without MMP
These can be extrapolated to simulate the effects of new transformations, fragments and modifications on the target protein.
Moreover, there has been a dramatic increase in information in public databases such as ChEMBL (a manually curated database of bioactive molecules with drug-like properties) and Pubchem (a database of chemical molecules and their activities against biological assays).
Their databases contain many structure–activity relationship (SAR) analyses, where MMPs with ML have been used to predict many bioactivity properties such as oral exposure, distribution coefficient (log D and log P), intrinsic clearance and ADMET.
Other Applications of AI in Drug Discovery
A different problem during secondary screening is assay interference caused by small molecules. Several approaches have been developed that allow scientists to flag potentially undruggable compounds, also known as “bad actors” or “nuisance compounds”. Usually, these compounds are aggregators, reactive compounds or pan-assay interference compounds (PAINS).
The solution to this problem comes from Hit Dexter. This recently introduced ML approach predicts how likely a small molecule is to trigger a false positive response in biochemical assays (including binding compounds based on “privileged scaffolds” to multiple binding sites).
The models used by Hit Dexter were derived from a dataset of 250,000 compounds with experimentally determined activity for at least 50 different protein groups.
The new Hit Dexter 2.0 web service covers both primary and secondary screening assays, providing user-friendly access to similarity-based methods for the prediction of aggregators and dark chemical matter (a set of drug-like compounds that has never shown bioactivity despite being extensively assayed), as well as a comprehensive collection of available rule sets for flagging frequent bad hitters and compounds including undesired substructures.
AI Startups Involved in Screening
While primary and secondary screening are considered part of early drug discovery, they play a critical role in drug development. The ability of AI to accelerate these studies means pharmaceutical companies can shave years and hundreds of millions of dollars off the process of bringing a drug to market. In turn, this translates to cheaper and more accessible medicines for the patients who require them.
Here are some startups that are bridging the gap between artificial intelligence and the drug screening process:

Metrion Biosciences (@Metrion_Biosci) is a UK-based contract research organization providing drug discovery services to pharmaceutical and bioscience customers that have just invested in a new High Throughput Screening capability and expanded facilities.
Delta 4 (@Delta4ai) is an Austrian digital drug discovery company that conducts in silico screening before experimental screening.

Micar Innovation (Micar21)(@Micar21Ltd) is a Bulgarian company focusing on the entire drug discovery process using improved structure based on in silico drug design with advanced activity and selectivity predictions and ADMET.

Synsight (@SynsightFR)is a French deep tech company developing a screening technology that enables the development of effective first-in-class drug candidates (for RNA targeting) based on a discovery platform of AI and cell imaging.

Remedium AI is a Canadian-based startup using ML techniques to address inefficiencies in drug screening. Its platform can analyze any protein with known or suspected therapeutic value with significantly greater accuracy than any comparable AI tool.

Phenomic (@PhenomicAI) is another Canadian-based biotech using advanced ML tools for process imaging, RNA sequencing and spatial transcriptomics data. It aims to understand the biology of single cells in complex multi-cell systems.

Aiforia (@aiforia_tech) is a Finnish company using images (tissues and cells) uploaded to their cloud to automate manual image analysis tasks and speed up workflows. Their cloud-based products cut down evaluation time from minutes to seconds, remove subjectivity in sample evaluation and allow for remote collaboration.

Variational AI (@VariationalAI) is a Canadian company specializing in a powerful new form of ML known as generative AI for drug discovery. It aims to free scientists from reliance on screening and libraries, both experimental and virtual, and to eventually generate de novo molecules with all the optimized properties to discover efficacious, safe, and synthesizable small molecule therapeutics in a fraction of the time and cost.
Reference
H.C. Stephen Chan, Hanbin Shan, Thamani Dahoun, HorstVogel, Shuguang Yuan, Advancing Drug Discovery via Artificial Intelligence (2019)
Amanda J. Minnich, Kevin McLoughlin, Margaret Tse, Jason Deng, Andrew Weber, Neha Murad, Benjamin D. Madej, Bharath Ramsundar, Tom Rush, Stacie Calad-Thomson, Jim Brase, and Jonathan E. Allen, AMPL: A Data-Driven Modeling Pipeline for Drug Discovery (2020)
About the Author

Marina is a molecular and cellular biologist, writer and a bit of an intellectual still trying to fit in. She has 20+ years of experience in academia, startups and MNCs. Currently is a life science consultant. She writes about A.I., drug discovery and biotech.